Processing and integrating PBMC TEA-seq data
Contents
Processing and integrating PBMC TEA-seq data#
This notebooks provides an example for TEA-seq data processing in Python.
TEA-seq is a method for trimodal single-cell profiling that allows to get gene expression, chromatin accessibility, and epitope information per cell. The method is described in Swanson et al., 2021.
The data used in this notebook is on peripheral blood mononuclear cells (PBMCs) and can be downloaded from GEO. We will use a single TEA-seq sample in this notebook — GSM5123951.
[1]:
# Change directory to the root folder of the repository
import os
os.chdir("../../")
Import libraries#
[2]:
import numpy as np
import pandas as pd
import scanpy as sc
[3]:
import muon as mu
import muon.atac as ac
import muon.prot as pt
[4]:
import matplotlib
from matplotlib import pyplot as plt
plt.rcParams['figure.dpi'] = 100
Prepare & load data#
We will use *_cellranger-arc_filtered_feature_bc_matrix.h5 and *_adt_counts.csv.gz files with counts. For chromatin accessibility, we’ll also use *_atac_filtered_fragments.tsv.gz files with fragments as well as metadata in *_atac_filtered_metadata.csv.gz.
For this notebook, we work with a single sample.
[5]:
# This is the directory where those files are downloaded to
data_dir = "data/teaseq"
[6]:
from glob import glob
for file in glob(f"{data_dir}/GSM5123951*"):
print(file)
data/teaseq/GSM5123951_X066-MP0C1W3_leukopak_perm-cells_tea_200M_atac_filtered_metadata.csv.gz
data/teaseq/GSM5123951_X066-MP0C1W3_leukopak_perm-cells_tea_200M_atac_filtered_fragments.tsv.gz.tbi
data/teaseq/GSM5123951_X066-MP0C1W3_leukopak_perm-cells_tea_48M_adt_counts.csv.gz
data/teaseq/GSM5123951_X066-MP0C1W3_leukopak_perm-cells_tea_200M_atac_filtered_fragments.tsv.gz
data/teaseq/GSM5123951_X066-MP0C1W3_leukopak_perm-cells_tea_200M_cellranger-arc_filtered_feature_bc_matrix.h5
Construct simple metadata parsed from the filenames:
[7]:
meta = {}
for file in glob(f"{data_dir}/GSM5123951*"):
tokens = os.path.basename(file).split("_")
meta[
tokens[0] # GSM5123951
] = tokens[1][-2:] # W3
meta
[7]:
{'GSM5123951': 'W3'}
First, we will load RNA+ATAC counts.
[8]:
get_h5_file = lambda root, s, w: f"{root}/{s}_X066-MP0C1{w}_leukopak_perm-cells_tea_200M_cellranger-arc_filtered_feature_bc_matrix.h5"
s, w = list(meta.items())[0]
mdata = mu.read_10x_h5(
get_h5_file(data_dir, s, w)
)
mdata.obs["sample"] = s
mdata.obs["well"] = w
mdata.update()
mdata.var_names_make_unique()
/usr/local/bin/miniconda3/envs/issue57/lib/python3.8/site-packages/anndata/_core/anndata.py:1830: UserWarning: Variable names are not unique. To make them unique, call `.var_names_make_unique`.
utils.warn_names_duplicates("var")
Added `interval` annotation for features from data/teaseq/GSM5123951_X066-MP0C1W3_leukopak_perm-cells_tea_200M_cellranger-arc_filtered_feature_bc_matrix.h5
/usr/local/bin/miniconda3/envs/issue57/lib/python3.8/site-packages/anndata/_core/anndata.py:1830: UserWarning: Variable names are not unique. To make them unique, call `.var_names_make_unique`.
utils.warn_names_duplicates("var")
mudata/_core/mudata.py:437: UserWarning: var_names are not unique. To make them unique, call `.var_names_make_unique`.
warnings.warn(
mudata/mudata/_core/mudata.py:437: UserWarning: var_names are not unique. To make them unique, call `.var_names_make_unique`.
warnings.warn(
[9]:
mdata
[9]:
MuData object with n_obs × n_vars = 7966 × 138155
obs: 'sample', 'well'
var: 'gene_ids', 'feature_types', 'genome', 'interval'
2 modalities
rna: 7966 x 36601
var: 'gene_ids', 'feature_types', 'genome', 'interval'
atac: 7966 x 101554
var: 'gene_ids', 'feature_types', 'genome', 'interval'We can use rich representation to explore the structure of the object.
[10]:
mu.set_options(display_style="html", display_html_expand=0b000);
[11]:
mdata
[11]:
Metadata.obs2 elements
| sample | object | GSM5123951,GSM5123951,GSM5123951,GSM5123951,... |
| well | object | W3,W3,W3,W3,W3,W3,W3,W3,W3,W3,W3,W3,W3,W3,W3,W3,... |
Embeddings & mappings.obsm0 elements
No embeddings & mappingsDistances.obsp0 elements
No distancesrna7966 × 36601
AnnData object 7966 obs × 36601 varLayers.layers0 elements
No layersMetadata.obs0 elements
No metadataEmbeddings.obsm0 elements
No embeddingsDistances.obsp0 elements
No distancesMiscellaneous.uns0 elements
No miscellaneousatac7966 × 101554
AnnData object 7966 obs × 101554 varLayers.layers0 elements
No layersMetadata.obs0 elements
No metadataEmbeddings.obsm0 elements
No embeddingsDistances.obsp0 elements
No distancesMiscellaneous.uns0 elements
No miscellaneousThen we will load protein counts.
[12]:
prot_counts = pd.read_csv(f"{data_dir}/{s}_X066-MP0C1{w}_leukopak_perm-cells_tea_48M_adt_counts.csv.gz")
prot_counts.set_index("cell_barcode", inplace=True)
prot_counts_total = prot_counts.total.values
del prot_counts["total"]
[13]:
prot_raw = mu.AnnData(X=prot_counts)
prot_raw.obs["total_counts"] = prot_counts_total
prot_raw.obs_names = prot_raw.obs_names + "-1"
prot_raw.var_names = "prot:" + prot_raw.var_names # feature names will be unambiguous then (e.g. gene or protein)
/var/folders/xt/tvy3s7w17vn1b700k_351pz00000gp/T/ipykernel_62585/1322958841.py:1: FutureWarning: X.dtype being converted to np.float32 from int64. In the next version of anndata (0.9) conversion will not be automatic. Pass dtype explicitly to avoid this warning. Pass `AnnData(X, dtype=X.dtype, ...)` to get the future behavour.
prot_raw = mu.AnnData(X=prot_counts)
[14]:
prot_raw
[14]:
AnnData object with n_obs × n_vars = 720873 × 46
obs: 'total_counts'
[15]:
prot_raw.var_names.values
[15]:
array(['prot:CD10', 'prot:CD11b', 'prot:CD11c', 'prot:CD123',
'prot:CD127', 'prot:CD14', 'prot:CD141', 'prot:CD16',
'prot:CD172a', 'prot:CD185', 'prot:CD19', 'prot:CD192',
'prot:CD197', 'prot:CD21', 'prot:CD24', 'prot:CD25', 'prot:CD269',
'prot:CD27', 'prot:CD278', 'prot:CD279', 'prot:CD3', 'prot:CD304',
'prot:CD319', 'prot:CD38', 'prot:CD39', 'prot:CD4', 'prot:CD40',
'prot:CD45RA', 'prot:CD45RO', 'prot:CD56', 'prot:CD66b',
'prot:CD71', 'prot:CD80', 'prot:CD86', 'prot:CD8a', 'prot:CD95',
'prot:FceRI', 'prot:HLA-DR', 'prot:IgD',
'prot:IgG1-K-Isotype-Control', 'prot:IgM', 'prot:KLRG1',
'prot:TCR-Va24-Ja18', 'prot:TCR-Va7.2', 'prot:TCR-a/b',
'prot:TCR-g/d'], dtype=object)
This is a 'raw' matrix, in which both cells and empty droplets are present. We will take advantage of this for denoising protein counts.
We can add a protein modality to our multimodal container. We will only add droplets with cells.
[16]:
mdata.mod["prot"] = prot_raw[mdata.obs_names].copy()
mdata.update()
[17]:
mdata
[17]:
Metadata.obs3 elements
| prot:total_counts | int64 | 863,957,1108,899,1155,835,820,643,822,776,406,... |
| sample | object | GSM5123951,GSM5123951,GSM5123951,GSM5123951,... |
| well | object | W3,W3,W3,W3,W3,W3,W3,W3,W3,W3,W3,W3,W3,W3,W3,W3,... |
Embeddings & mappings.obsm0 elements
No embeddings & mappingsDistances.obsp0 elements
No distancesrna7966 × 36601
AnnData object 7966 obs × 36601 varLayers.layers0 elements
No layersMetadata.obs0 elements
No metadataEmbeddings.obsm0 elements
No embeddingsDistances.obsp0 elements
No distancesMiscellaneous.uns0 elements
No miscellaneousatac7966 × 101554
AnnData object 7966 obs × 101554 varLayers.layers0 elements
No layersMetadata.obs0 elements
No metadataEmbeddings.obsm0 elements
No embeddingsDistances.obsp0 elements
No distancesMiscellaneous.uns0 elements
No miscellaneousprot7966 × 46
AnnData object 7966 obs × 46 varLayers.layers0 elements
No layersMetadata.obs1 element
| total_counts | int64 | 863,957,1108,899,1155,835,820,643,822,776,406,... |
Embeddings.obsm0 elements
No embeddingsDistances.obsp0 elements
No distancesMiscellaneous.uns0 elements
No miscellaneousProcessing individual modalities#
Protein modality (epitopes)#
[18]:
prot = mdata["prot"]
DSB normalisation#
This normalisation method developed for CITE-seq data uses background droplets defined by low RNA content in order to estimate background protein signal and remove it from the data. More details and its original implementation are available in the ``dsb` GitHub repository <https://github.com/niaid/dsb>`__.
Preserve original counts in a layer before the normalisation:
[19]:
prot.layers['counts'] = prot.X
Specify features corresponding to the isotype controls:
[20]:
isotypes = prot.var_names.values[["Isotype" in v for v in prot.var_names]]
print(isotypes)
['prot:IgG1-K-Isotype-Control']
Normalize counts with dsb:
[21]:
pt.pp.dsb(mdata, prot_raw, isotype_controls=isotypes, random_state=1)
muon/muon/_prot/preproc.py:109: UserWarning: empty_counts_range values are not provided, treating all the non-cells as empty droplets
warn(
Plot values to visualise the effect of normalisation:
[22]:
sc.pl.scatter(mdata['prot'], x="prot:CD3", y="prot:CD19", layers='counts')
sc.pl.scatter(mdata['prot'], x="prot:CD3", y="prot:CD19")
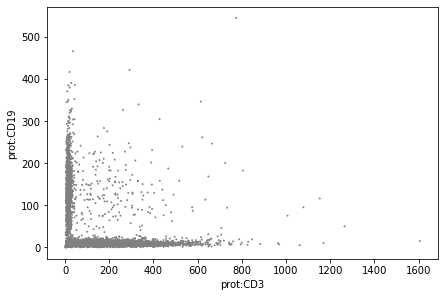
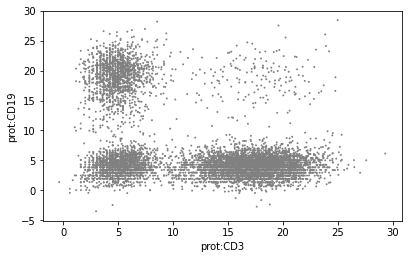
Downstream analysis#
We can run conventional methods like PCA on the normalised protein counts:
[23]:
sc.tl.pca(prot)
[24]:
sc.pl.pca(prot, color=['prot:CD19', 'prot:CD11b'])
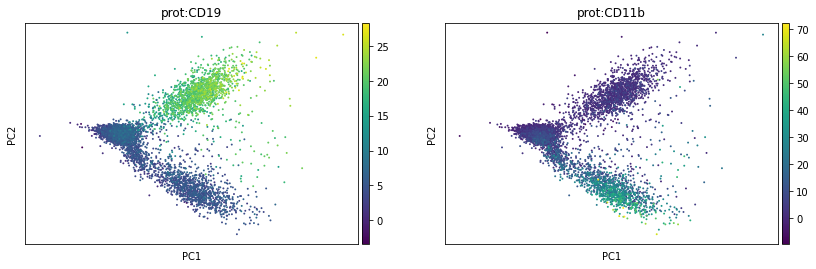
[25]:
sc.pp.neighbors(prot)
sc.tl.umap(prot, random_state=1)
[26]:
sc.pl.umap(prot, color=['prot:CD3', 'prot:CD19', 'prot:CD11b'])

RNA modality#
[27]:
rna = mdata.mod['rna']
rna
[27]:
AnnData object with n_obs × n_vars = 7966 × 36601
var: 'gene_ids', 'feature_types', 'genome', 'interval'
QC#
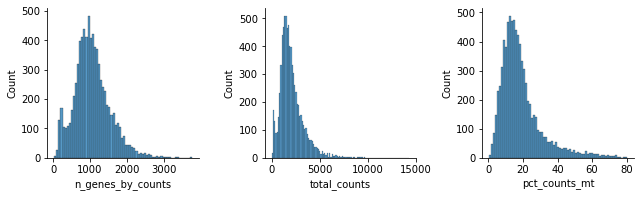
Perform some quality control. For now, we will filter out cells that do not pass QC from the whole dataset.
[28]:
rna.var['mt'] = rna.var_names.str.startswith('MT-') # annotate the group of mitochondrial genes as 'mt'
sc.pp.calculate_qc_metrics(rna, qc_vars=['mt'], percent_top=None, log1p=False, inplace=True)
[29]:
mu.pl.histogram(rna, ['n_genes_by_counts', 'total_counts', 'pct_counts_mt'])
Filter genes which expression is not detected:
[30]:
mu.pp.filter_var(rna, 'n_cells_by_counts', lambda x: x >= 10) # gene detected at least in 10 cells
Filter cells:
[31]:
mu.pp.filter_obs(rna, 'n_genes_by_counts', lambda x: (x >= 200) & (x < 2500))
mu.pp.filter_obs(rna, 'total_counts', lambda x: (x > 500) & (x < 5000))
mu.pp.filter_obs(rna, 'pct_counts_mt', lambda x: x < 30)
Let’s see how the data looks after filtering:
[32]:
mu.pl.histogram(rna, ['n_genes_by_counts', 'total_counts', 'pct_counts_mt'])
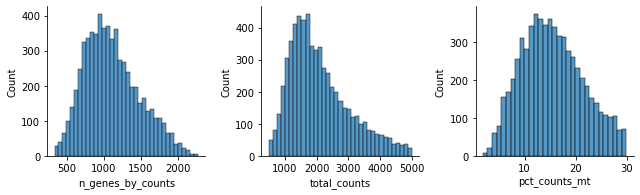
Normalisation#
We’ll normalise the data so that we get log-normalised counts to work with.
[33]:
sc.pp.normalize_total(rna, target_sum=1e4)
[34]:
sc.pp.log1p(rna)
Feature selection#
We will label highly variable genes that we’ll use for downstream analysis.
[35]:
sc.pp.highly_variable_genes(rna, min_mean=0.02, max_mean=4, min_disp=0.5)
[36]:
sc.pl.highly_variable_genes(rna)
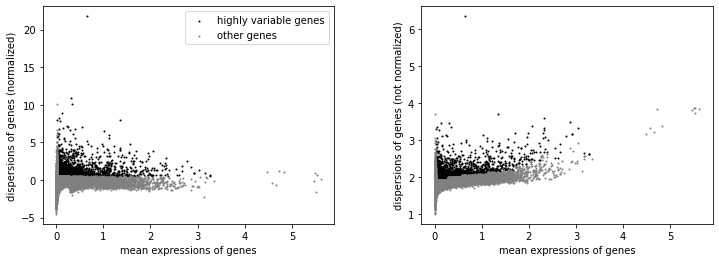
[37]:
np.sum(rna.var.highly_variable)
[37]:
2910
Scaling#
We’ll save log-normalised counts in the "lognorm" layer:
[38]:
rna.layers["lognorm"] = rna.X.copy()
… and scale the log-normalised counts to zero mean and unit variance:
[39]:
sc.pp.scale(rna, max_value=10)
Downstream analysis#
Having filtered low-quality cells, normalised the counts matrix, and performed feature selection, we can already use this data for multimodal integration.
It is usually a good idea to study individual modalities first. Moreover, some multimodal methods can utilize principal components space or cell neighbourhood graphs from individual modalities.
Below we run PCA on the scaled matrix, compute cell neighbourhood graph, and perform clustering to define cell types.
PCA and neighbourhood graph#
[40]:
sc.tl.pca(rna, svd_solver='arpack')
To visualise the result, we will use some markers for (large-scale) cell populations we expect to see such as T cells and NK cells (CD2), B cells (CD79A), and KLF4 (monocytes).
[41]:
sc.pl.pca(rna, color=['CD2', 'CD3E', 'NKG7', 'KLF4', 'MS4A1', 'CD79A', 'IRF8'], layer="lognorm")
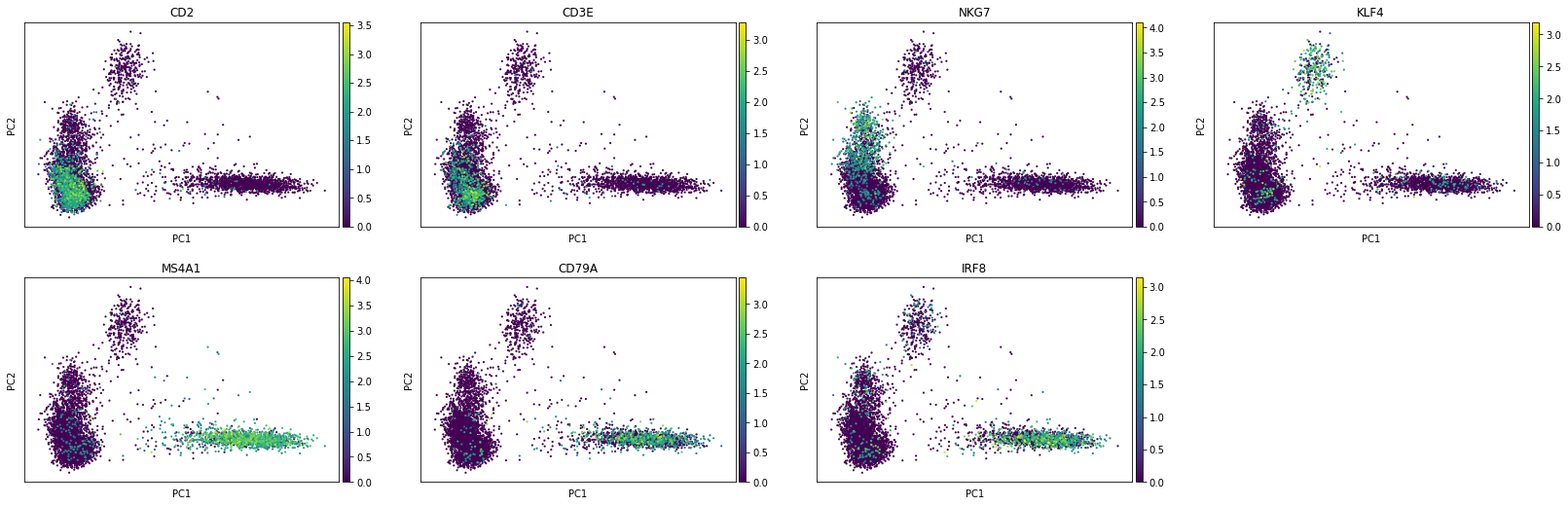
The first principal component (PC1) is separating T/NK (left), myeloid (middle), and B (right) cells while monocyte-related features seem to drive the second one. Also we see plasmocytoid dendritic cells (marked by IRF8) being close to B cells in the PC1/PC2 space.
[42]:
sc.pl.pca_variance_ratio(rna, log=True)
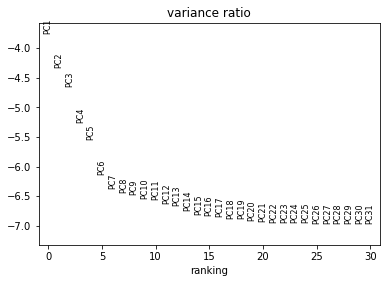
Now we can compute a neighbourhood graph for cells:
[43]:
sc.pp.neighbors(rna, n_neighbors=10, n_pcs=20)
Non-linear dimensionality reduction and clustering#
With the neighbourhood graph computed, we can now perform clustering. We will use leiden clustering as an example.
[44]:
sc.tl.leiden(rna, resolution=.75)
To visualise the results, we’ll first generate a 2D latent space with cells that we can colour according to their cluster assignment.
[45]:
sc.tl.umap(rna, spread=1., min_dist=.5, random_state=11)
[46]:
sc.pl.umap(rna, color="leiden", legend_loc="on data")
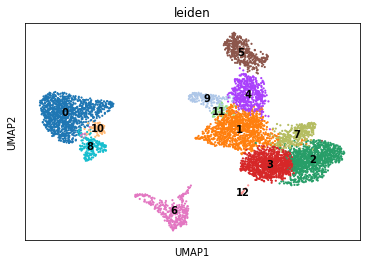
Plotting across modalities#
With mu.pl.embedding interface we can display an embedding from an individual modality (e.g. 'rna') and colour cells by a feature (variable) from another modality.
While variables names should be unique across all modalities, all individual modalities as well as the mdata object itself can have e.g. UMAP basis computed. To point to a basis from a specific modality, use mod:basis syntax, e.g. with "rna:X_umap" in the example below mdata.mod['rna'].obsm["X_umap"] basis is going to be used.
[48]:
mu.pl.embedding(mdata, basis="rna:X_umap", color=["prot:IgD", "prot:IgM", "IGHD", "IGHM", "prot:CD127", "IL7R"], layer={"rna": "lognorm"}, ncols=3)

ATAC modality#
[49]:
atac = mdata['atac']
atac
[49]:
AnnData object with n_obs × n_vars = 7966 × 101554
var: 'gene_ids', 'feature_types', 'genome', 'interval'
Locate the fragments file (use tabix -p bed to generate an index file for it):
[50]:
ac.tl.locate_fragments(atac, f"{data_dir}/{s}_X066-MP0C1{w}_leukopak_perm-cells_tea_200M_atac_filtered_fragments.tsv.gz")
Adding metadata to the ATAC modality:
[51]:
metadata = pd.read_csv(f"{data_dir}/{s}_X066-MP0C1{w}_leukopak_perm-cells_tea_200M_atac_filtered_metadata.csv.gz")
pd.set_option('display.max_columns', 500)
metadata.head()
[51]:
| original_barcodes | n_fragments | n_duplicate | n_mito | n_unique | altius_count | altius_frac | gene_bodies_count | gene_bodies_frac | peaks_count | peaks_frac | tss_count | tss_frac | barcodes | cell_name | well_id | chip_id | batch_id | pbmc_sample_id | DoubletScore | DoubletEnrichment | TSSEnrichment | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | GTGTGAGCATGCTATG-1 | 38760 | 17870 | 2 | 17316 | 13692 | 0.790714 | 11785 | 0.680584 | 10043 | 0.579984 | 5100 | 0.294525 | 69c6c7c43ef411ebb8b442010a19c80f | unblacked_accurate_myotis | X066-AP0C1W3 | X066-AP0C1 | X066 | X066-Well3 | 0.000000 | 0.257143 | 8.046 |
| 1 | TCACCGGCACAGAACG-1 | 12820 | 7294 | 12 | 4291 | 3751 | 0.874155 | 3128 | 0.728968 | 3159 | 0.736192 | 2194 | 0.511303 | 69c7d3ee3ef411ebb8b442010a19c80f | fabulous_panphobic_tahr | X066-AP0C1W3 | X066-AP0C1 | X066 | X066-Well3 | 0.000000 | 0.114286 | 8.997 |
| 2 | AAAGGACGTACAATGT-1 | 16117 | 9524 | 0 | 4989 | 4561 | 0.914211 | 3602 | 0.721988 | 3863 | 0.774303 | 2828 | 0.566847 | 69be295c3ef411ebb8b442010a19c80f | tricky_anaemic_mayfly | X066-AP0C1W3 | X066-AP0C1 | X066 | X066-Well3 | 0.000000 | 0.085714 | 11.081 |
| 3 | CAGCCTAAGACAACAG-1 | 44967 | 24156 | 0 | 17456 | 15918 | 0.911893 | 12033 | 0.689333 | 13724 | 0.786205 | 7241 | 0.414814 | 69c161123ef411ebb8b442010a19c80f | agonizing_haughty_collie | X066-AP0C1W3 | X066-AP0C1 | X066 | X066-Well3 | 2.351825 | 1.828571 | 12.804 |
| 4 | ACTCGCTTCCAGGAAA-1 | 16526 | 8896 | 0 | 6227 | 5584 | 0.896740 | 4286 | 0.688293 | 4747 | 0.762325 | 2658 | 0.426851 | 69bf6b003ef411ebb8b442010a19c80f | exorcistic_harmonic_bushbaby | X066-AP0C1W3 | X066-AP0C1 | X066 | X066-Well3 | 0.000000 | 1.400000 | 21.016 |
[52]:
atac.obs = atac.obs.join(metadata.set_index("original_barcodes"))
Please note this metadata is available for the filtered (after QC) ATAC data so this modality can be just filtered using these cells.
For demonstration purposes we will still go through processing steps before removing barcodes that were filtered out.
QC#
Perform some quality control filtering out cells with too few peaks and peaks detected in too few cells. For now, we will filter out cells that do not pass QC.
[53]:
sc.pp.calculate_qc_metrics(atac, percent_top=None, log1p=False, inplace=True)
[54]:
mu.pl.histogram(atac, ['total_counts', 'n_genes_by_counts'])
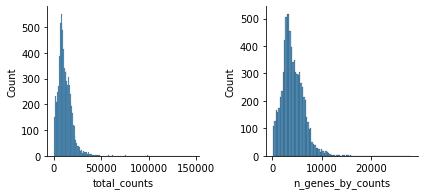
Filter peaks which expression is not detected:
[55]:
mu.pp.filter_var(atac, 'n_cells_by_counts', lambda x: x >= 5) # a peak is detected in 5 cells or more
Filter cells:
[56]:
mu.pp.filter_obs(atac, 'total_counts', lambda x: (x >= 1000) & (x <= 30000))
mu.pp.filter_obs(atac, 'n_genes_by_counts', lambda x: (x >= 500) & (x <= 10000)) # number of peaks per cell
Let’s see how the data looks after filtering:
[57]:
mu.pl.histogram(atac, ['total_counts', 'n_genes_by_counts'])
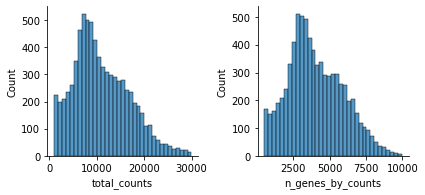
There are a few expectations about how ATAC-seq data looks like as noted in the hitchhiker’s guide to ATAC-seq data analysis for instance.
Nucleosome signal#
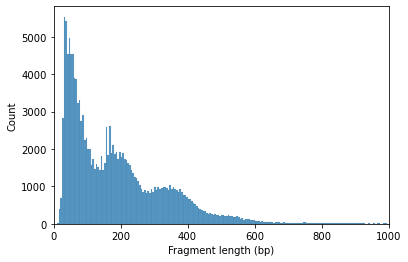
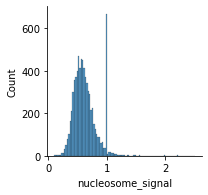
Fragment size distribution typically reflects nucleosome binding pattern showing enrichment around values corresponding to fragments bound to a single nucleosome (between 147 bp and 294 bp) as well as nucleosome-free fragments (shorter than 147 bp).
[58]:
ac.pl.fragment_histogram(atac, region='chr1:1-2000000', barcodes="barcodes")
Fetching Regions...: 100%|███████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 1/1 [00:00<00:00, 1.19it/s]
The ratio of mono-nucleosome cut fragments to nucleosome-free fragments can be called nucleosome signal, and it can be estimated using a subset of fragments.
[59]:
ac.tl.nucleosome_signal(atac, n=1e6, barcodes="barcodes")
Reading Fragments: 100%|█████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 1000000/1000000 [00:06<00:00, 149011.41it/s]
[60]:
mu.pl.histogram(atac, "nucleosome_signal", kde=False)
TSS enrichment#
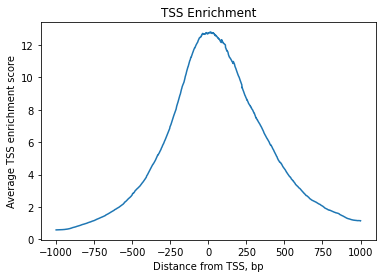
We can expect chromatin accessibility enriched around transcription start sites (TSS) compared to accessibility of flanking regions. Thus this measure averaged across multiple genes can serve as one more quality control metric.
The positions of transcription start sites can be obtained from the interval field of the gene annotation in the rna modality:
[61]:
ac.tl.get_gene_annotation_from_rna(mdata['rna']).head(3) # accepts MuData with 'rna' modality or mdata['rna'] AnnData directly
[61]:
| Chromosome | Start | End | gene_id | gene_name | |
|---|---|---|---|---|---|
| AL627309.5 | chr1 | 149706 | 173862 | ENSG00000241860 | AL627309.5 |
| LINC01409 | chr1 | 778757 | 803934 | ENSG00000237491 | LINC01409 |
| LINC01128 | chr1 | 827597 | 860227 | ENSG00000228794 | LINC01128 |
TSS enrichment function will return an AnnData object with cells x bases dimensions where bases correspond to positions around TSS and are defined by extend_upstream and extend_downstream parameters, each of them being 1000 bp by default. It will also record tss_score in the original object.
[62]:
tss = ac.tl.tss_enrichment(mdata, n_tss=1000, barcodes="barcodes") # by default, features=ac.tl.get_gene_annotation_from_rna(mdata)
Fetching Regions...: 100%|█████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 1000/1000 [00:16<00:00, 61.24it/s]
[63]:
tss
[63]:
AnnData object with n_obs × n_vars = 7639 × 2001
obs: 'n_fragments', 'n_duplicate', 'n_mito', 'n_unique', 'altius_count', 'altius_frac', 'gene_bodies_count', 'gene_bodies_frac', 'peaks_count', 'peaks_frac', 'tss_count', 'tss_frac', 'barcodes', 'cell_name', 'well_id', 'chip_id', 'batch_id', 'pbmc_sample_id', 'DoubletScore', 'DoubletEnrichment', 'TSSEnrichment', 'n_genes_by_counts', 'total_counts', 'nucleosome_signal', 'tss_score'
var: 'TSS_position'
[64]:
ac.pl.tss_enrichment(tss)
Now we’ll remove the barcodes that were filtered out as indicated in the provided metadata.
[65]:
# To avoid issues with nan being converted to 'nan' when the column is categorical,
# we explicitely convert it to str
mu.pp.filter_obs(atac, atac.obs.barcodes.astype(str) != 'nan')
Normalisation#
[66]:
# Save original counts
atac.layers["counts"] = atac.X
There can be multiple options for ATAC-seq data normalisation such as LSI that uses TF-IDF transformation.
Here we will use the same log-normalisation and PCA that we are used to from scRNA-seq analysis.
[67]:
sc.pp.normalize_per_cell(atac, counts_per_cell_after=1e4)
sc.pp.log1p(atac)
Feature selection#
We will label highly variable peaks that we’ll use for downstream analysis.
[68]:
sc.pp.highly_variable_genes(atac, min_mean=0.05, max_mean=1.5, min_disp=.5)
[69]:
sc.pl.highly_variable_genes(atac)
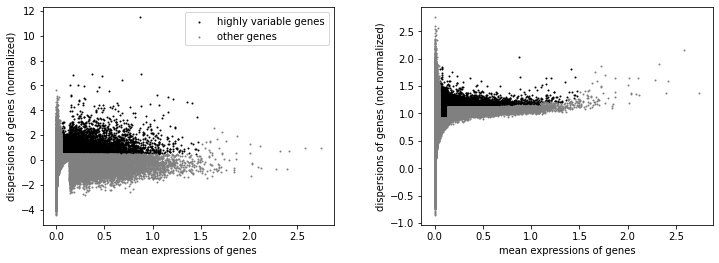
[70]:
np.sum(atac.var.highly_variable)
[70]:
10867
Scaling#
We’ll save log-transformed counts in the "lognorm" layer:
[71]:
atac.layers["lognorm"] = atac.X.copy()
Downstream analysis#
PCA#
For this notebook, we are using PCA on the log-normalised counts in atac.X as described above.
[72]:
sc.pp.scale(atac)
sc.tl.pca(atac)
We can only colour our plots by cut counts in individual peaks with scanpy:
[73]:
sc.pl.pca(atac, color=["n_genes_by_counts", "n_counts"], layer="lognorm")
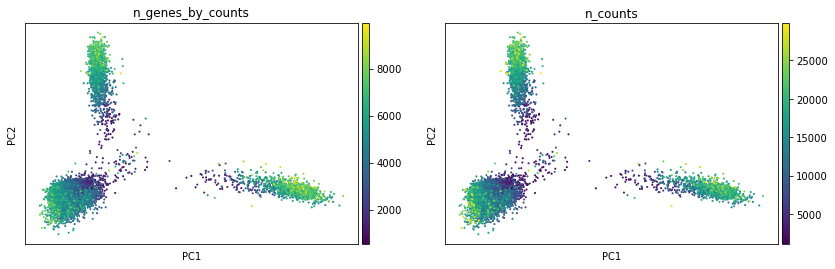
Now we will compute a neighbourhood graph for cells that we’ll use for clustering later on.
[74]:
sc.pp.neighbors(atac, n_neighbors=10, n_pcs=30)
Non-linear dimensionality reduction and clustering#
To stay comparable to the gene expression notebook, we will use leiden to cluster cells.
[75]:
sc.tl.leiden(atac, resolution=.5)
We’ll use UMAP latent space for visualisation below.
[76]:
sc.tl.umap(atac, spread=1.5, min_dist=.5, random_state=30)
[77]:
sc.pl.umap(atac, color=["leiden", "n_genes_by_counts"], legend_loc="on data")

Multi-omics analyses#
For the downstream multimodal analysis, we will use only cells passing QC steps in each of the modality. Since cells were filtered out in each modality above, we can just take their intersection:
[78]:
mu.pp.intersect_obs(mdata)
Multiplex clustering#
We can run clustering based on neighbours information from each modality taking advantage of multiplex versions of algorithms such as leiden or louvain.
[79]:
mdata.uns = dict()
[80]:
mu.tl.leiden(mdata, resolution=[1., 1., 1.], random_state=1, key_added="leiden_multiplex")
[85]:
mu.pl.embedding(mdata, basis="rna:X_umap", color=["prot:CD3", "leiden_multiplex", "rna:leiden"])
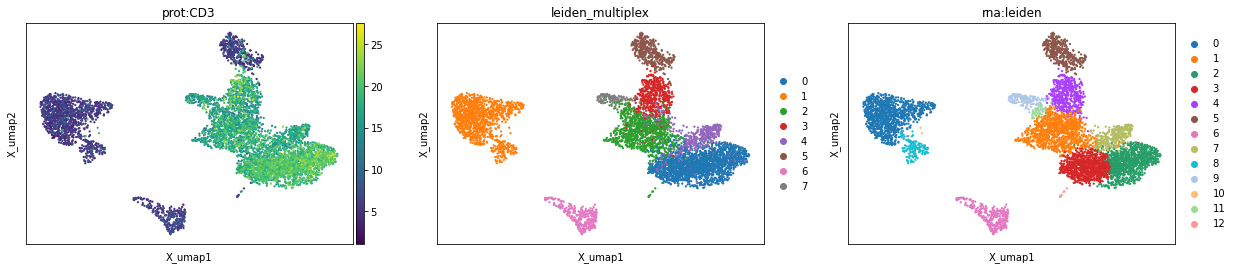
Multi-omics factor analysis#
To generate an interpretable latent space for all three modalities, we will now run multi-omic factor analysis — a group factor analysis method that will allow us to learn an interpretable latent space jointly on both modalities. Intuitively, it can be viewed as a generalisation of PCA for multi-omics data. More information about this method can be found on the MOFA website.
[86]:
prot.var["highly_variable"] = True
mdata.update()
[88]:
mu.tl.mofa(mdata, outfile="models/pbmc_w3_teaseq.hdf5")
#########################################################
### __ __ ____ ______ ###
### | \/ |/ __ \| ____/\ _ ###
### | \ / | | | | |__ / \ _| |_ ###
### | |\/| | | | | __/ /\ \_ _| ###
### | | | | |__| | | / ____ \|_| ###
### |_| |_|\____/|_|/_/ \_\ ###
### ###
#########################################################
Loaded view='rna' group='group1' with N=5805 samples and D=2910 features...
Loaded view='atac' group='group1' with N=5805 samples and D=10867 features...
Loaded view='prot' group='group1' with N=5805 samples and D=46 features...
Model options:
- Automatic Relevance Determination prior on the factors: True
- Automatic Relevance Determination prior on the weights: True
- Spike-and-slab prior on the factors: False
- Spike-and-slab prior on the weights: True
Likelihoods:
- View 0 (rna): gaussian
- View 1 (atac): gaussian
- View 2 (prot): gaussian
######################################
## Training the model with seed 1 ##
######################################
Converged!
#######################
## Training finished ##
#######################
Warning: Output file models/pbmc_w3_teaseq.hdf5 already exists, it will be replaced
Saving model in models/pbmc_w3_teaseq.hdf5...
Saved MOFA embeddings in .obsm['X_mofa'] slot and their loadings in .varm['LFs'].
This adds X_mofa embeddings to the .obsm slot of the MuData object itself. The weights for the latent factors are stored in .varm["LFs"].
[89]:
mu.pl.mofa(mdata, color=['prot:CD3', 'prot:CD14'])
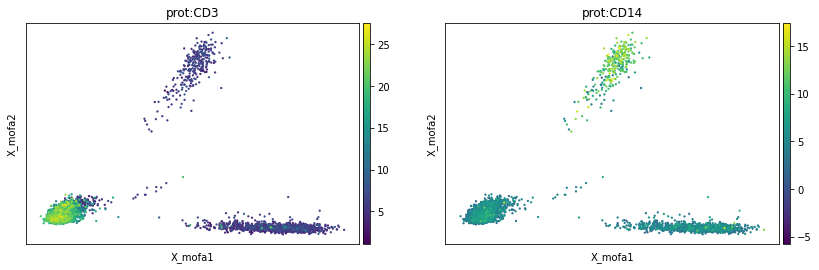
MOFA factors can be used to define cell neighbourhood graphs, which are thus based on all the modalities, as well as for subsequent non-linear latent space construction such as UMAP and clustering.
[90]:
sc.pp.neighbors(mdata, use_rep="X_mofa", key_added='mofa')
sc.tl.umap(mdata, min_dist=.2, random_state=1, neighbors_key='mofa')
[130]:
sc.tl.leiden(mdata, resolution=.3, neighbors_key='mofa', key_added='leiden_mofa')
[131]:
mu.pl.umap(mdata, color=['leiden_mofa'], frameon=False, title="UMAP(MOFA) embedding")
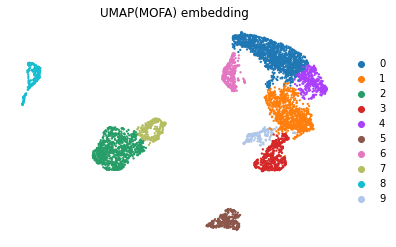
[93]:
mu.pl.umap(mdata, color=['CD3E', 'prot:CD3'], layer={"rna": "lognorm"}, frameon=False, cmap='viridis')
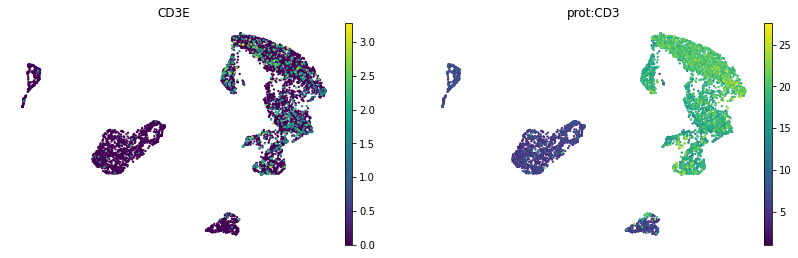
[95]:
mdata.obsm["X_mofa_umap"] = mdata.obsm["X_umap"].copy()
Interpreting the model#
We can describe factors by the top features, which they are defined by.
[96]:
mu.pl.mofa_loadings(mdata)


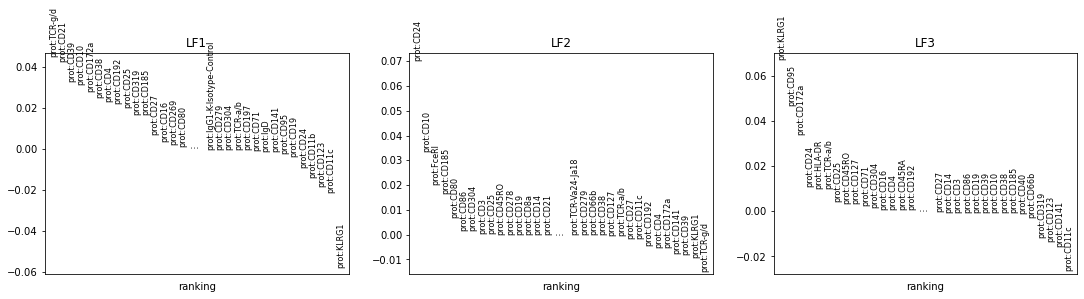
More detailed analysis of the MOFA model can be done with mofax, which is available here, using the model file saved during the training above.
Weighted nearest neighbours#
Another way to leverage multimodal information is with the weighted nearest neighbours (WNN) method, which constructs a multimodal cell neighbourhood graph based on cell neighbourhood graphs of individual modalities. muon implements WNN as it is described in `Hao et al., 2020 <>`__ with the generalisation to an arbitrary number of modalities proposed in Swanson et al., 2021.
For this to work, we’ll have to re-calculate neighbourhood in each modality because we have filtered out cells after that had been done above. That usually leads to having cells with no neighbours, and muon will be explicit about it and will return an error.
[97]:
for m in mdata.mod.keys():
sc.pp.neighbors(mdata[m])
[98]:
mu.pp.neighbors(mdata, key_added='wnn')
Again, we can use the defined cell neighbours to perform clustering or non-linear dimensionality reduction.
[145]:
sc.tl.leiden(mdata, resolution=.55, neighbors_key='wnn', key_added='leiden_wnn')
[148]:
mu.tl.umap(mdata, random_state=10, neighbors_key='wnn')
[149]:
mu.pl.umap(mdata, color="leiden_wnn")

[150]:
mdata.obsm["X_wnn_umap"] = mdata.obsm["X_umap"].copy()
Cell types annotation#
We can use the results of any multiple multimodal analyses — or multiple ones — to define cell types. Alternatively, cell types can be defined using a single modality, e.g. 'prot', and the cell types labels robustness can then be checked with multimodal embeddings.
[151]:
mu.pl.embedding(mdata, basis="X_wnn_umap", color=list(map(
lambda x: "prot:" + x,
[
"CD4", "CD45RA", "CD45RO", # CD4 T cells
"CD8a", # CD8 T cells
"KLRG1", # NK cells
"CD14", "CD16", # monocytes
"CD19", "CD21", "IgD", # B cells
]
)))

Label clusters according to the markers:
[155]:
new_cluster_names = {
"0": "naïve CD4+ T", "1": "naïve B", "2": "memory CD4+ T",
"4": "naïve CD8+ T", "3": "effector CD8+ T", "5": "CD16 mono",
"6": "CD14 mono", "7": "active B", "8": "NK",
}
mdata.obs['celltype'] = mdata.obs.leiden_wnn.astype("str").values
mdata.obs.celltype = mdata.obs.celltype.astype("category")
mdata.obs.celltype = mdata.obs.celltype.cat.rename_categories(new_cluster_names)
[156]:
mdata.obs.celltype.cat.reorder_categories([
'naïve CD4+ T', 'memory CD4+ T', 'NK', 'effector CD8+ T', 'naïve CD8+ T',
'naïve B', 'active B',
'CD14 mono', 'CD16 mono',
], inplace=True)
Add colours:
[157]:
cmap = plt.get_cmap('rainbow')
colors = cmap(np.linspace(0, 1, len(mdata.obs.celltype.cat.categories)))
mdata.uns["celltype_colors"] = list(map(matplotlib.colors.to_hex, colors))
[158]:
mu.pl.umap(mdata, color="celltype", frameon=False, title="UMAP(WNN)")

Saving multimodal data on disk#
[159]:
mdata.update()
mdata
[159]:
Metadata.obs42 elements
| rna:n_genes_by_counts | int32 | 1375,957,1052,1253,783,681,922,828,1083,1161,998,... |
| rna:total_counts | float32 | 2509.00,1405.00,1777.00,2399.00,1501.00,1018.00,... |
| rna:total_counts_mt | float32 | 200.00,119.00,298.00,277.00,448.00,144.00,411.00,... |
| rna:pct_counts_mt | float32 | 7.97,8.47,16.77,11.55,29.85,14.15,24.49,27.18,... |
| rna:leiden | object | 2,2,7,0,8,3,8,3,5,1,0,2,2,2,1,2,6,0,3,0,0,4,4,1,1,... |
| atac:n_fragments | float64 | 13705.00,11567.00,25610.00,30969.00,16007.00,... |
| atac:n_duplicate | float64 | 8269.00,6897.00,15043.00,17513.00,8686.00,6703.00,... |
| atac:n_mito | float64 | 50.00,0.00,0.00,128.00,899.00,4.00,3.00,0.00,0.00,... |
| atac:n_unique | float64 | 4163.00,3516.00,8395.00,10883.00,4839.00,3152.00,... |
| atac:altius_count | float64 | 3777.00,3145.00,7613.00,9992.00,4314.00,2806.00,... |
| atac:altius_frac | float64 | 0.91,0.89,0.91,0.92,0.89,0.89,0.90,0.89,0.89,0.91,... |
| atac:gene_bodies_count | float64 | 3070.00,2545.00,6168.00,7951.00,3514.00,2263.00,... |
| atac:gene_bodies_frac | float64 | 0.74,0.72,0.73,0.73,0.73,0.72,0.71,0.72,0.74,0.74,... |
| atac:peaks_count | float64 | 3245.00,2648.00,6466.00,8669.00,3617.00,2408.00,... |
| atac:peaks_frac | float64 | 0.78,0.75,0.77,0.80,0.75,0.76,0.77,0.74,0.74,0.76,... |
| atac:tss_count | float64 | 2422.00,2053.00,5054.00,6165.00,2644.00,1812.00,... |
| atac:tss_frac | float64 | 0.58,0.58,0.60,0.57,0.55,0.57,0.54,0.57,0.59,0.57,... |
| atac:barcodes | object | 69be0ea43ef411ebb8b442010a19c80f,... |
| atac:cell_name | object | viceregal_unconstant_hornet,... |
| atac:well_id | object | X066-AP0C1W3,X066-AP0C1W3,X066-AP0C1W3,... |
| atac:chip_id | object | X066-AP0C1,X066-AP0C1,X066-AP0C1,X066-AP0C1,... |
| atac:batch_id | object | X066,X066,X066,X066,X066,X066,X066,X066,X066,X066,... |
| atac:pbmc_sample_id | object | X066-Well3,X066-Well3,X066-Well3,X066-Well3,... |
| atac:DoubletScore | float64 | 0.00,0.00,0.00,0.00,0.00,11.71,0.00,0.83,0.00,... |
| atac:DoubletEnrichment | float64 | 0.37,1.23,1.26,0.69,0.89,2.46,0.34,1.69,1.00,0.74,... |
| atac:TSSEnrichment | float64 | 17.69,18.75,21.54,21.58,15.49,23.57,19.00,18.32,... |
| atac:n_genes_by_counts | int32 | 2846,2366,4987,6409,3105,2126,3849,2472,3107,4512,... |
| atac:total_counts | float32 | 7103.00,5916.00,14559.00,19267.00,8280.00,5259.00,... |
| atac:nucleosome_signal | float64 | 0.67,0.77,0.69,0.42,0.54,0.69,0.53,0.52,0.41,0.69,... |
| atac:tss_score | float64 | 9.91,5.17,6.09,4.33,6.03,10.30,5.83,3.20,14.67,... |
| atac:n_counts | float32 | 7102.00,5915.00,14559.00,19266.00,8280.00,5259.00,... |
| atac:leiden | object | 0,0,7,1,9,4,9,0,6,2,1,0,0,0,2,0,3,1,0,1,1,5,2,2,4,... |
| prot:total_counts | int64 | 863,957,899,835,820,643,822,776,406,923,909,1320,... |
| sample | object | GSM5123951,GSM5123951,GSM5123951,GSM5123951,... |
| well | object | W3,W3,W3,W3,W3,W3,W3,W3,W3,W3,W3,W3,W3,W3,W3,W3,... |
| leiden_multiplex | object | 0,0,4,1,1,0,1,0,5,3,1,0,0,0,2,0,6,1,0,1,1,3,4,2,2,... |
| leiden_mofa | category | 0,0,6,2,7,0,7,0,5,3,2,4,0,0,1,4,8,7,0,7,2,3,3,1,1,... |
| rna:mod_weight | float64 | 0.33,0.30,0.34,0.30,0.30,0.33,0.21,0.32,0.33,0.31,... |
| atac:mod_weight | float64 | 0.33,0.30,0.33,0.30,0.26,0.35,0.29,0.33,0.34,0.31,... |
| prot:mod_weight | float64 | 0.35,0.40,0.33,0.40,0.45,0.33,0.50,0.36,0.33,0.39,... |
| leiden_wnn | category | 0,0,4,1,7,0,7,0,5,3,1,0,0,0,2,0,6,1,0,1,1,3,3,2,2,... |
| celltype | category | naïve CD4+ T,naïve CD4+ T,naïve CD8+ T,naïve B,... |
Embeddings & mappings.obsm4 elements
| X_mofa | float64 | numpy.ndarray | 10 dims |
| X_umap | float32 | numpy.ndarray | 2 dims |
| X_mofa_umap | float32 | numpy.ndarray | 2 dims |
| X_wnn_umap | float32 | numpy.ndarray | 2 dims |
Distances.obsp4 elements
| mofa_distances | float64 | scipy.sparse._csr.csr_matrix |
| mofa_connectivities | float32 | scipy.sparse._csr.csr_matrix |
| wnn_distances | float32 | scipy.sparse._csr.csr_matrix |
| wnn_connectivities | float32 | scipy.sparse._csr.csr_matrix |
Miscellaneous.uns7 elements
| leiden | dict | 1 element | params: resolution,random_state,n_iterations |
| mofa | dict | 3 elements | connectivities_key,distances_key,params |
| umap | dict | 1 element | params: a,b,random_state |
| leiden_mofa_colors | list | 11 elements | #1f77b4,#ff7f0e,#279e68,#d62728,#aa40fc,#8c564b,... |
| wnn | dict | 3 elements | connectivities_key,distances_key,params |
| leiden_wnn_colors | list | 11 elements | #1f77b4,#ff7f0e,#279e68,#d62728,#aa40fc,#8c564b,... |
| celltype_colors | list | 9 elements | #8000ff,#4062fa,#00b5eb,#40ecd4,#80ffb4,#c0eb8d,... |
rna5805 × 16381
AnnData object 5805 obs × 16381 varLayers.layers1 element
| lognorm | float32 | scipy.sparse._csr.csr_matrix |
Metadata.obs5 elements
| n_genes_by_counts | int32 | 1375,957,1052,1253,783,681,922,828,1083,1161,998,... |
| total_counts | float32 | 2509.00,1405.00,1777.00,2399.00,1501.00,1018.00,... |
| total_counts_mt | float32 | 200.00,119.00,298.00,277.00,448.00,144.00,411.00,... |
| pct_counts_mt | float32 | 7.97,8.47,16.77,11.55,29.85,14.15,24.49,27.18,... |
| leiden | category | 2,2,7,0,8,3,8,3,5,1,0,2,2,2,1,2,6,0,3,0,0,4,4,1,1,... |
Embeddings.obsm2 elements
| X_pca | float32 | numpy.ndarray | 50 dims |
| X_umap | float32 | numpy.ndarray | 2 dims |
Distances.obsp2 elements
| distances | float64 | scipy.sparse._csr.csr_matrix |
| connectivities | float32 | scipy.sparse._csr.csr_matrix |
Miscellaneous.uns7 elements
| log1p | dict | 1 element | base: None |
| hvg | dict | 1 element | flavor: seurat |
| pca | dict | 3 elements | params,variance,variance_ratio |
| neighbors | anndata.compat._overloaded_dict.OverloadedDict | 3 elements | connectivities_key,distances_key,params |
| leiden | dict | 1 element | params: resolution,random_state,n_iterations |
| umap | dict | 1 element | params: a,b,random_state |
| leiden_colors | list | 13 elements | #1f77b4,#ff7f0e,#279e68,#d62728,#aa40fc,#8c564b,... |
atac5805 × 96760
AnnData object 5805 obs × 96760 varLayers.layers2 elements
| counts | float32 | scipy.sparse._csr.csr_matrix | |
| lognorm | float32 | scipy.sparse._csr.csr_matrix |
Metadata.obs27 elements
| n_fragments | float64 | 13705.00,11567.00,25610.00,30969.00,16007.00,... |
| n_duplicate | float64 | 8269.00,6897.00,15043.00,17513.00,8686.00,6703.00,... |
| n_mito | float64 | 50.00,0.00,0.00,128.00,899.00,4.00,3.00,0.00,0.00,... |
| n_unique | float64 | 4163.00,3516.00,8395.00,10883.00,4839.00,3152.00,... |
| altius_count | float64 | 3777.00,3145.00,7613.00,9992.00,4314.00,2806.00,... |
| altius_frac | float64 | 0.91,0.89,0.91,0.92,0.89,0.89,0.90,0.89,0.89,0.91,... |
| gene_bodies_count | float64 | 3070.00,2545.00,6168.00,7951.00,3514.00,2263.00,... |
| gene_bodies_frac | float64 | 0.74,0.72,0.73,0.73,0.73,0.72,0.71,0.72,0.74,0.74,... |
| peaks_count | float64 | 3245.00,2648.00,6466.00,8669.00,3617.00,2408.00,... |
| peaks_frac | float64 | 0.78,0.75,0.77,0.80,0.75,0.76,0.77,0.74,0.74,0.76,... |
| tss_count | float64 | 2422.00,2053.00,5054.00,6165.00,2644.00,1812.00,... |
| tss_frac | float64 | 0.58,0.58,0.60,0.57,0.55,0.57,0.54,0.57,0.59,0.57,... |
| barcodes | object | 69be0ea43ef411ebb8b442010a19c80f,... |
| cell_name | object | viceregal_unconstant_hornet,... |
| well_id | category | X066-AP0C1W3,X066-AP0C1W3,X066-AP0C1W3,... |
| chip_id | category | X066-AP0C1,X066-AP0C1,X066-AP0C1,X066-AP0C1,... |
| batch_id | category | X066,X066,X066,X066,X066,X066,X066,X066,X066,X066,... |
| pbmc_sample_id | category | X066-Well3,X066-Well3,X066-Well3,X066-Well3,... |
| DoubletScore | float64 | 0.00,0.00,0.00,0.00,0.00,11.71,0.00,0.83,0.00,... |
| DoubletEnrichment | float64 | 0.37,1.23,1.26,0.69,0.89,2.46,0.34,1.69,1.00,0.74,... |
| TSSEnrichment | float64 | 17.69,18.75,21.54,21.58,15.49,23.57,19.00,18.32,... |
| n_genes_by_counts | int32 | 2846,2366,4987,6409,3105,2126,3849,2472,3107,4512,... |
| total_counts | float32 | 7103.00,5916.00,14559.00,19267.00,8280.00,5259.00,... |
| nucleosome_signal | float64 | 0.67,0.77,0.69,0.42,0.54,0.69,0.53,0.52,0.41,0.69,... |
| tss_score | float64 | 9.91,5.17,6.09,4.33,6.03,10.30,5.83,3.20,14.67,... |
| n_counts | float32 | 7102.00,5915.00,14559.00,19266.00,8280.00,5259.00,... |
| leiden | category | 0,0,7,1,9,4,9,0,6,2,1,0,0,0,2,0,3,1,0,1,1,5,2,2,4,... |
Embeddings.obsm2 elements
| X_pca | float32 | numpy.ndarray | 50 dims |
| X_umap | float32 | numpy.ndarray | 2 dims |
Distances.obsp2 elements
| distances | float64 | scipy.sparse._csr.csr_matrix |
| connectivities | float32 | scipy.sparse._csr.csr_matrix |
Miscellaneous.uns8 elements
| files | dict | 1 element | fragments: data/teaseq/GSM5123951_X066-MP0C1W3_leukopak_perm-cells_tea_200M_atac_filtered_fragments.tsv.gz |
| log1p | dict | 1 element | base: None |
| hvg | dict | 1 element | flavor: seurat |
| pca | dict | 3 elements | params,variance,variance_ratio |
| neighbors | anndata.compat._overloaded_dict.OverloadedDict | 3 elements | connectivities_key,distances_key,params |
| leiden | dict | 1 element | params: resolution,random_state,n_iterations |
| umap | dict | 1 element | params: a,b,random_state |
| leiden_colors | list | 11 elements | #1f77b4,#ff7f0e,#279e68,#d62728,#aa40fc,#8c564b,... |
prot5805 × 46
AnnData object 5805 obs × 46 varLayers.layers1 element
| counts | float32 | numpy.ndarray |
Metadata.obs1 element
| total_counts | int64 | 863,957,899,835,820,643,822,776,406,923,909,1320,... |
Embeddings.obsm2 elements
| X_pca | float32 | numpy.ndarray | 45 dims |
| X_umap | float32 | numpy.ndarray | 2 dims |
Distances.obsp2 elements
| distances | float64 | scipy.sparse._csr.csr_matrix |
| connectivities | float32 | scipy.sparse._csr.csr_matrix |
Miscellaneous.uns3 elements
| pca | dict | 3 elements | params,variance,variance_ratio |
| neighbors | anndata.compat._overloaded_dict.OverloadedDict | 3 elements | connectivities_key,distances_key,params |
| umap | dict | 1 element | params: a,b,random_state |
We will now write mdata object to an .h5mu file.
[163]:
mdata.write(f"{data_dir}/pbmc_w3_teaseq.h5mu", compression="gzip")